die gemeinnützige Paul-Martini-Stiftung, kontrolliert von Betrugs Pharma Firmen
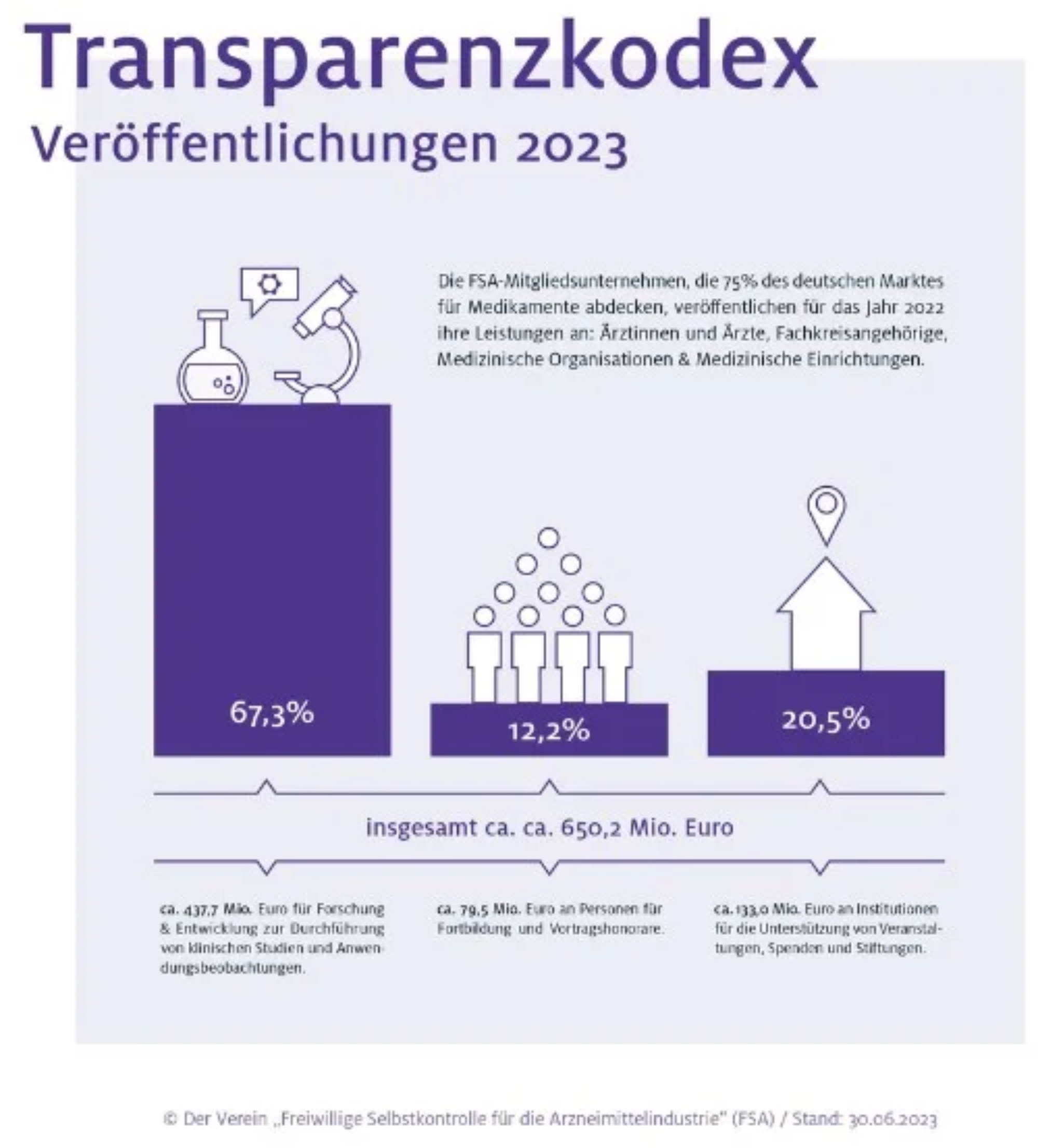
650 Millionen Zuwendungen der Arzneimittelindustrie an ÄrztInnen, Organisationen und Einrichtungen
Das berichtet für das Jahr 2022 der Verein Freiwillige Selbstkontrolle für die Arzneimittelindustrie e. V. (FSA),
»Die FSA-Mitgliedsuntemehmen, die 75% des deutschen Marktes für Medikamente abdecken, veröffentlichen für das Jahr 2022 ihre Leistungen an Ärztinnen und Ärzte, Fachkreisangehörige, Medizinische Organisationen & Medizinische Einrichtungen.«

Über die gemeinnützige Paul-Martini-Stiftung
Wer vertritt diese Stiftung, die nach einem Faschisten und Antisemiten der ersten Stunde benannt ist und die mit den Koryphäen der Corona-Maßnahmen am 17./18.11 eine Tagung zu diesem Thema veranstaltet (s. hier)? Nicht nur ihr Vorstandssprecher ist als „Medizinischer Direktor bei Pfizer in Deutschland“ ein unmittelbarer Vertreter der Pharmaindustrie. Der komplette Vorstand besteht aus Konzernvertretern.


„Über die gemeinnützige Paul-Martini-Stiftung“ weiterlesen
Wenn 48 Pharmaunternehmen ein COVID-Symposium sponsern…
Unter diesem Titel ist am 1.11.23 diese Pressemitteilung auf mezis.de erschienen (hier mit einem Nachtrag zum Namenspatron der Stiftung – „nicht ohne antisemitische Vorbehalte„):

»MEZIS fordert Leopoldina auf, sich als Mitveranstalterin zurückzuziehen
Das aktuell geplante COVID-Symposium der Paul-Martini-Stiftung am 17. und 18. November 2023 in der Kaiserin-Friedrich-Stiftung in Berlin: „Prävention und Therapie von COVID-19: Update und Learnings“ ist in punkto Industrienähe und Interessenkonflikte extrem:
-
- Die als Veranstalter firmierende Paul-Martini-Stiftung wird direkt und vollständig von 48 Unternehmen der Pharmaindustrie und damit von den direkten Nutznießern des Abends finanziert.
- „Wenn 48 Pharmaunternehmen ein COVID-Symposium sponsern…“ weiterlesen
„Mir wäre ein gebrochenes Bein lieber“
Dieses Zitat eines Betroffenen findet man in einem Artikel auf vdi-nachrichten.com vom 2.11.23 (Bezahlschranke).

Danach
»… verzeichnete [die Deutsche Psychotherapeutenvereinigung (DPtV)] allein während der Coronapandemie einen Anstieg der Nachfrage in den Praxen um 40 %. Für die kommenden Jahre prognostiziere das Zentralinstitut für die kassenärztliche Versorgung eine nochmals steigende Nachfrage um 25 %, heißt es bei der DPtV…«
Die Mitherausgeberin des aktuellen AOK-Fehlzeiten-Reports stellt fest: „“Mir wäre ein gebrochenes Bein lieber““ weiterlesen
650 Millionen Zuwendungen der Arzneimittelindustrie an ÄrztInnen, Organisationen und Einrichtungen
Das berichtet für das Jahr 2022 der Verein Freiwillige Selbstkontrolle für die Arzneimittelindustrie e. V. (FSA),
»Die FSA-Mitgliedsuntemehmen, die 75% des deutschen Marktes für Medikamente abdecken, veröffentlichen für das Jahr 2022 ihre Leistungen an Ärztinnen und Ärzte, Fachkreisangehörige, Medizinische Organisationen & Medizinische Einrichtungen.«
Über die gemeinnützige Paul-Martini-Stiftung
Wer vertritt diese Stiftung, die nach einem Faschisten und Antisemiten der ersten Stunde benannt ist und die mit den Koryphäen der Corona-Maßnahmen am 17./18.11 eine Tagung zu diesem Thema veranstaltet (s. hier)? Nicht nur ihr Vorstandssprecher ist als „Medizinischer Direktor bei Pfizer in Deutschland“ ein unmittelbarer Vertreter der Pharmaindustrie. Der komplette Vorstand besteht aus Konzernvertretern.
„Über die gemeinnützige Paul-Martini-Stiftung“ weiterlesen
Wenn 48 Pharmaunternehmen ein COVID-Symposium sponsern…
Unter diesem Titel ist am 1.11.23 diese Pressemitteilung auf mezis.de erschienen (hier mit einem Nachtrag zum Namenspatron der Stiftung – „nicht ohne antisemitische Vorbehalte„):
»MEZIS fordert Leopoldina auf, sich als Mitveranstalterin zurückzuziehen
Das aktuell geplante COVID-Symposium der Paul-Martini-Stiftung am 17. und 18. November 2023 in der Kaiserin-Friedrich-Stiftung in Berlin: „Prävention und Therapie von COVID-19: Update und Learnings“ ist in punkto Industrienähe und Interessenkonflikte extrem:
-
- Die als Veranstalter firmierende Paul-Martini-Stiftung wird direkt und vollständig von 48 Unternehmen der Pharmaindustrie und damit von den direkten Nutznießern des Abends finanziert.
- „Wenn 48 Pharmaunternehmen ein COVID-Symposium sponsern…“ weiterlesen
„Mir wäre ein gebrochenes Bein lieber“
Dieses Zitat eines Betroffenen findet man in einem Artikel auf vdi-nachrichten.com vom 2.11.23 (Bezahlschranke).
Danach
»… verzeichnete [die Deutsche Psychotherapeutenvereinigung (DPtV)] allein während der Coronapandemie einen Anstieg der Nachfrage in den Praxen um 40 %. Für die kommenden Jahre prognostiziere das Zentralinstitut für die kassenärztliche Versorgung eine nochmals steigende Nachfrage um 25 %, heißt es bei der DPtV…«
Die Mitherausgeberin des aktuellen AOK-Fehlzeiten-Reports stellt fest: „“Mir wäre ein gebrochenes Bein lieber““ weiterlesen
Hinterlasse einen Kommentar
Neueste Kommentare
| infomarinadurrescom bei 84,4 % der Migranten sind Verb… | |
| balkansurfer bei Kriegs Verbrechen der Amerikan… | |
| lupodemaregmailcom bei ChatGPT, KI will die unfähige… | |
| lupodemaregmailcom bei Vorsatz und ohne jede Grundlag… | |
| balkansurfer bei Vorsatz und ohne jede Grundlag… | |
| balkansurfer bei Vorsatz und ohne jede Grundlag… | |
| lupodemaregmailcom bei Robert Habeck im Mafia Stile,… | |
| infomarinadurrescom bei Ursula von der Leyen: Lügen ha… | |
| balkansurfer bei Der NATO, Pentagon, Angela Mer… | |
| balkansurfer bei Der Schwindel mit dem Fisch:… |

Veröffentlicht am von Illa
Der nicht zugelassene „Impfstoff“ für Milliarden
Genaugenommen hat der „Impfstoff“ von BioNTech/Pfizer (im folgenden Text: B/P), der Milliarden von Menschen teilweise mehrmals injiziert wurde und mit dem Milliarden Euro verdient wurden, keine Zulassung. Die bedingte (in der EU über die EMA) bzw. Notfallzulassung (in den USA über die FDA) im Dezember 2020 wurden für die Präparate vergeben, die mit dem „Prozess 1“ aus der Kleinproduktion (35 ml) für die klinischen Versuche hergestellt worden waren. Das fundamental andere Herstellungsverfahren im „Prozess 2“ mit dem über tausendfachen Volumen führte allerdings zu einem völlig anderen Produkt — was sogar mit bloßem Auge zu sehen war: Durch „Prozess 1“ entstanden Präparate, die „frei von sichtbaren Partikeln“ waren, während es danach nur noch hieß: „Im wesentlichen frei von sichtbaren Partikeln“.
Die Großproduktion verwendet das Darmbakterium Escherichia coli (E.coli), in das zusätzlich zur bakteriellen DNA ein speziell hergestelltes ringförmiges DNA-Plasmid eingebracht wurde, von dem die modRNA transkribiert wird. Das bei beiden Prozessen gleichermaßen als „In Vitro Transciption“ bezeichnete Verfahren zur modRNA-Herstellung wurde im „Prozess 1“ dagegen unter Laborbedingungen mittels RT-PCR durchgeführt, an dem keine E.coli beteiligt waren. Im „Prozess 2“ waren in der Folge sowohl mehr sichtbare Verunreinigungen als auch labortechnisch nachweisbare Fremdstoffe vorhanden.
B/P sollten lediglich nachträglich beweisen, dass beide Prozesse zu gleichen Ergebnissen führen, wobei es im wesentlichen um die Integrität der modRNA ging, also darum, ob diese im selben Umfang vollständig vorliegt oder ob es mit „Prozess 2“ mehr unvollständige Sequenzen gab als bei „Prozess 1“. Letzteres war der Fall, durfte aber nicht sein und führte zu „Blotgate“ (s.u.). Ein weiterer Unterschied zwischen „Prozess 1“ und „Prozess 2“ liegt in der Plasmid-DNA aus E.coli, die also nur bei den Präparaten aus der Großproduktion in die Lipid-Nanopartikeln gelangt und zusammen mit der modRNA in das Zellinnere geschleust wird, wie Anfang des Jahres in den USA bekannt wurde. Hierzulande wurde es September, bis die Nachricht bei einer recht überschaubaren Öffentlichkeit ankam.
„Prozess 1“
Ebenfalls im September erschien der Preprint über die „Forensische Analyse der 38 Todesfälle in dem 6‑Monats-Zwischenbericht der klinischen Studie des Pfizer/BioNTech BNT162b2 mRNA Impfstoffs“. Sie wurde vom „DailyClout Pfizer/BioNTech Documents Investigations Team“ vorgelegt, einem Teil der „Gruppe von etwa 3500 Medizinern, Wissenschaftlern, Datenanalysten, Statistikern, Juristen und weiteren Personen“, die die „Impfstoff“-Daten analysiert haben, die die FDA freigeben mußte, statt sie wie geplant jahrzehntelang unter Verschluss zu halten. Hier ging es um die vom 27. Juli 2020 (Versuchsbeginn für Phase 2/3 zur Bestimmung von „Wirksamkeit und Sicherheit“) bis zum 13. März 2021 (Daten-Ende für den Interim-Bericht) gemeldeten insgesamt 38 Todesfälle.
An diesem klinischen Versuch hatten ca. 44.000 Menschen teilgenommen, jeweils die Hälfte in der Wirkstoff- und Placebogruppe (im folgenden Text: W und P). Bei der Abgabe der Daten für den Zulassungsantrag an die FDA am 14.11.2020 meldeten B/P 6 Todesfälle (2 W und 4 P). Am 20.11. wurde der Zulassungsantrag mit diesen Daten gestellt, am 10.12. fand das abschließende Treffen zwischen FDA und B/P statt und am 11.12. erhielt „Comirnaty“ die Notfallzulassung in den USA. Damit endete die „Verblindete Placebo-kontrollierte Periode“ und die „Unverblindete Periode“ begann, in der alle Teilnehmer erfuhren, ob sie Wirkstoff oder Placebo erhalten hatten und das Angebot erhielten, sich „impfen“ zu lassen, was viele wahrnahmen. Die Kontrollgruppe wurde abgeschafft.
Bis zum abschließenden Treffen am 10.12. waren B/P insgesamt 16 Todesfälle nachgemeldet worden (jeweils 8 in beiden Gruppen), davon waren 4 W und 1 P im Zeitraum bis zum 14.11. verstorben. Bis zur Datenabgabe an diesem Tag gab es damit insgesamt 6 (statt 2) Todesfälle bei W und 5 (statt 4) bei P, was von B/P gegenüber der Zulassungsbehörde nicht korrigiert wurde. Eine kardiovaskuläre Todesursache wurde in dieser Gruppe bei 4 W und 2 P ausgemacht; die Autoren errechneten bei der Analyse aller 38 Verstorbenen eine 3,7fache Erhöhung in der Anzahl der Todesfälle durch kardiovaskuläre Ereignisse in W verglichen mit P. Das auch schon im Dezember erkennbare Sicherheitssignal wurde von B/P vertuscht und erst von der FDA sowie nachfolgend von der EMA und anderen Zulassungsbehörden weltweit ignoriert.
Selbst das Produkt aus dem vergleichsweise überschaubaren „Prozess 1“ erforderte also erhebliche Anstrengungen, die in dem Artikel ausführlicher beschrieben wurden, um nach außen hin eine Zulassung nicht sofort verdächtig erscheinen zu lassen. Dazu gehörten außerdem u.a. laut der Whistleblowerin Brook Jackson die Vertuschung von Verstößen gegen Vorschriften und die Umdeklaration von Schädigungen durch die „Impfung“ wie bei Augusto Roux , der eine Myokarditis erlitt. Selbst der „gute“ B/P‑Stoff war schlecht.
„Prozess 1“ und „Prozess 2“
Auf der Website von Pfizer ist das Protokoll für die klinischen Zulassungsversuche zu finden, in das am 6.10. folgender Absatz eingefügt wurde:
„6.1.1. Herstellungsprozess
Der Umfang der BNT162b2-Herstellung wurde erhöht, um das künftige Angebot zu unterstützen. BNT162b2, das mit dem Herstellungsverfahren hergestellt wurde, das eine erhöhte Versorgung ermöglicht (‚Prozess 2‘), wird in der Studie an etwa 250 Teilnehmer im Alter von 16 bis 55 Jahren pro Charge verabreicht. Die Sicherheit und Immunogenität von prophylaktischem BNT162b2 bei Personen im Alter von 16 bis 55 Jahren, die mit Material geimpft wurden, das mit dem bestehenden Herstellungsverfahren ‚Prozess 1‘ hergestellt wurde, und mit Material aus Chargen, die mit dem Herstellungsverfahren zur Unterstützung eines erhöhten Angebots, ‚Prozess 2‘, hergestellt wurden, werden beschrieben.
Kurz gesagt betreffen die Prozessänderungen die Produktionsmethode für die DNA-Vorlage, von der die RNA-Arzneimittelsubstanz transkribiert wird, und das Verfahren zur Reinigung der RNA-Arzneimittelsubstanz. Das BNT162b2-Arzneimittel wird dann mit einem hochskalierten LNP-Herstellungsverfahren produziert.“
Selbst der EMA war aufgefallen, dass dadurch ein Problem entsteht, wie aus nachträglich publik gewordenen Unterlagen hervorging:
„Nach Rücksprache mit dem Antragsteller wurde bestätigt, dass DP[Drug Product]-Chargen, die aus frühen Prozess-2-Chargen mit geringerer RNA-Integrität hergestellt wurden, vor kurzem in klinische Studien eingeführt wurden. Da jedoch der Stichtag für die klinische Zwischenanalyse (IA) geändert wurde, enthält die IA keine Daten von Probanden, die mit Prozess-2-Material verabreicht wurden, und das Unternehmen geht nicht davon aus, dass Prozess 2 in den Datensatz der endgültigen Analyse aufgenommen wird.“
Es gab folglich keine klinischen Daten für „Prozess 2“ im Zulassungsverfahren und die biochemischen Unterschiede wurden weitestmöglich negiert. Der Kriminologe Josh Guetzkow und der Mathematiker Retsef Levi schrieben an das British Medical Journal: „Zu den Unterschieden gehören Änderungen an der DNA-Vorlage, die zur Transkription der RNA und der Reinigungsphase verwendet wird, sowie am Herstellungsprozess der Lipid-Nanopartikel. Insbesondere wurde gezeigt, dass ‚Prozess 2‘-Chargen eine wesentlich geringere mRNA-Integrität aufwiesen.“ Dies war das Thema von „Blotgate“ mit den von B/T manipulierten Ergebnissen, die mittels der Labormethode Western Blot produziert wurden. Das Vorhandensein eines höheren Anteils nicht-intakter modRNA in der Großproduktion sollte dadurch möglichst wenig auffallen und führte zur Einreichung von Blot-Fotos bei der EMA, mit denen offensichtlich etwas nicht stimmte, die aber ihren Zweck erfüllten; dokumentiert ist dies in „BioNTech, Pfizer und Blotgate“.
Guetzkow und Levi schrieben weiter: „Erkenntnisse aus vorhandenen Forschungs- und Versuchsdokumenten unterstreichen die Bedeutung der öffentlichen Offenlegung der Analyse zum Vergleich der Reaktogenität und Sicherheit der Chargen von Prozess 1 und 2, wie im Versuchsprotokoll angegeben, sowie allgemeinerer Chargendaten auf Patientenebene aus der Studie.“ Kurz gesagt: „Der Prozess ist das Produkt“ und dieses „Produkt 2“ ist niemals zugelassen worden.